The Computational Population Genetics Group, or CpG group, is located in the International Laboratory for Human Genome Research at the Campus Juriquilla of the National Autonomous University of Mexico. We develop new analytical theory and create new computational methods to infer the impact of past evolutionary processes on patterns of genetic and phenotypic variation. We are particularly interested on pursuing three research lines:
Understanding the joint impact of demographic history and natural selection in patterns of genetic variation
The efficiency of natural selection to remove deleterious mutations and to increase the frequency of beneficial mutations is dependent on the past population history. In the past, we have studied how population size decreases in canids has changed the amount of deleterious genetic variation across the genome (Marsden CD*, Ortega-Del Vecchyo D*, et al. 2016 PNAS; Robinson et al. 2016 Genome Research). These analysis included the development of a new software program, PReFerSim (Ortega-Del Vecchyo D et al. 2016 Bioinformatics), that can perform simulations of independent genetic variants under natural selection in different demographic scenarios. Recently, we have became interested on developing new methods to infer the impact of natural selection acting on new mutations using informative features of the data that have not been leveraged by previous studies. This interest motivated us to create a method that uses haplotypic information to infer the distribution of fitness effects for new mutations (Ortega-Del Vecchyo et al. 2022 Genetics). The efforts in this line of research are very active and these are some of the projects we are pursuing:
– Alan is developing a new method to infer the distribution of fitness effects using information from the Ancestral Recombination Graph.
– Alessandro is creating a method to estimate the impact of natural selection acting on new variants by jointly analyzing ancient and present-day samples.
Our key collaborators on the projects delineated here are Christian Huber, Charleston Chiang, Parul Johri, Bernard Y. Kim, María Ávila-Arcos and Federico Sánchez-Quinto.
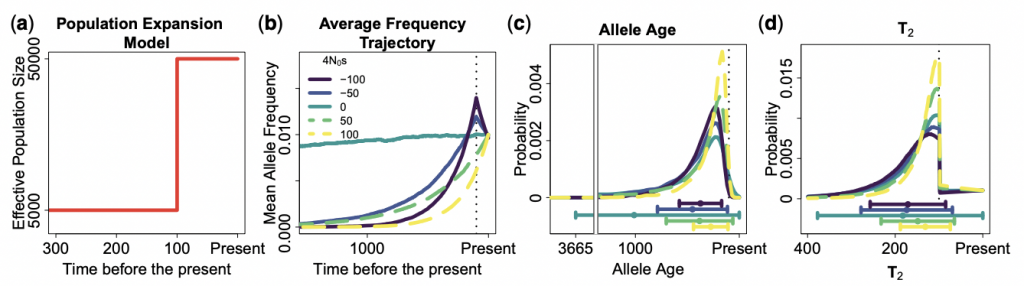
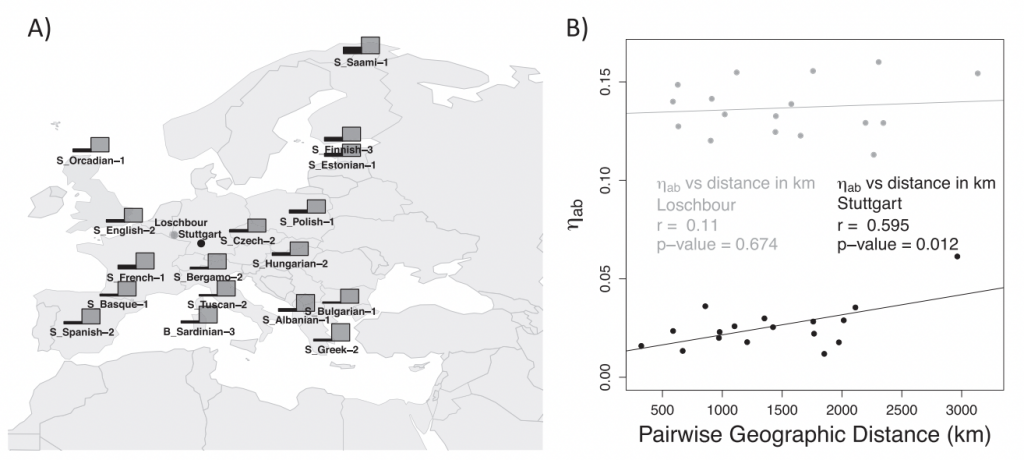
Inferring past demographic history using present-day and ancient samples
We are currently working on developing methods and theory to infer past population history by analyzing ancient and present-day samples. In the past, we have developed analytical theory to study how the use of ancient samples changes measures of population differentiation under different demographic scenarios (Ortega-Del Vecchyo and Slatkin 2019 Heredity) and have also developed an ABC approach to co-estimate homoplasy metrics and past population history (Ortega-Del Vecchyo et al. 2017 BMC Evolutionary Biology). We have also developed new metrics to detect segments of the genome that are the product of introgression (Lopez-Fang et al 2024 PLoS Genetics) and have also collaborated on projects to analyze the past demographic history of pine species (Figueroa-Corona et al. 2022 Ecology and Evolution). Some of our ongoing efforts include the following projects:
– Liliana is developing methods that take into account the contamination and sequencing errors found in ancient samples to infer past population history.
– David Peede, a collaborator and PhD student advised Emilia Huerta-Sánchez, is developing new metrics to estimate the proportion of introgressed segments in a genome.
Our key collaborators on this topic are David Peede and Emilia Huerta-Sánchez.
Complex trait evolution
We are interested in understanding how major evolutionary processes interact to shape complex trait variation. In the past we have published a review on what factors determine the spatial structure of complex traits (Sohail*, Izarraras-Gomez* and Ortega-Del Vecchyo 2021, GBE) and have also analyzed how natural selection hampers our ability to predict traits in the past (Añorve-Garibay et al. 2025, AJHG). Some of the current projects of the lab in this line of research include:
– Valeria Cabrera is analyzing how directional selection drives changes in patterns of genetic and phenotypic variation.
– Andrea is analyzing how natural selection impacts patterns of genetic variation.
Our key collaborators on this line of research are Mashaal Sohail, Charleston Chiang, Christian Huber and Emilia Huerta-Sánchez.